
What is EDS?
Ehlers-Danlos Syndromes are a group of connective tissue disorders that can be inherited and are varied both in how affect the body and in their genetic causes. They are generally characterized by joint hypermobility (joints that stretch further than normal), skin hyperextensibility (skin that can be stretched further than normal), and tissue fragility.
ehlers-danlos.com
What type do you have?
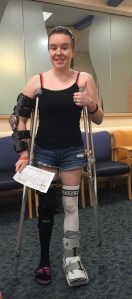
I have hEDS (Hypermobility EDS) or formerly called Stage III. I started showing symptoms at 10, but wasn’t diagnosed until college. In 2015, I started having severe joint subluxations and dislocations and was diagnosed. The joints/areas that I currently have tendon snapping, subluxations, or dislocations are found in almost every area of my body: cervical instability, Right Patella, L achilles, R Soleus, R & L Ankle + Talus, R & L Tibial Rotations, R Fibia, L Hip, R Hip, R Scapula, L Scapula, R Shoulder, L Shoulder, R Elbow, L elbow, L wrist, Fingers and Thumbs.
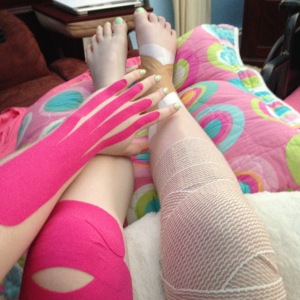 Unless I am in the acute healing phase of a injury (i.e. sublux, dislocation, ligament/tendon tear, etc.) almost of my braces are used on an as needed base. I use them when I feel like a joint is having a bad day or is loose, or of course if something has popped out. The only braces that I wear daily are my R knee brace with HKAFO (only when I am going out somewhere I typically do not wear it around the house unless there is an active dislocation.) I wear my neck brace and have support pillows for my shoulders for every car ride; I also have my shoulders, scapulas, lower leg and ankle taped 90% of the time.
Unless I am in the acute healing phase of a injury (i.e. sublux, dislocation, ligament/tendon tear, etc.) almost of my braces are used on an as needed base. I use them when I feel like a joint is having a bad day or is loose, or of course if something has popped out. The only braces that I wear daily are my R knee brace with HKAFO (only when I am going out somewhere I typically do not wear it around the house unless there is an active dislocation.) I wear my neck brace and have support pillows for my shoulders for every car ride; I also have my shoulders, scapulas, lower leg and ankle taped 90% of the time.
There are not many days that you will find me without more than one brace on and/or KT tape holding something together. I also demonstrate almost all of the symptoms of EDS
Fun Fact: I have enough braces to make up about 3 1/2 people!
What is hEDS?
Hypermobile EDS as yet has no identified distinctive cause; it evolves over time. The “hypermobility” phase (the first years of life) involves contortionism and propensity for sprains and dislocations; pain, often limited to lower limbs, also with fine motor or repetitive tasks; easy fatiguability may be a feature, together with voiding dysfunction. The “pain” phase (starting 2nd to 4th decade of life) includes more widespread and progressively worsening musculoskeletal pain, pelvic pain in women, and headache; exacerbation of fatigue; and is often associated with additional complaints. The “stiffness” phase (observed in a few adults and elderly only) results in general reduction of joint mobility; significant reduction in functionality due to disabling symptoms of pain and fatigue, and limitations from reduced muscle mass and weakness, prior injuries, and arthritis. The clinical description of hEDS in the medical literature has expanded considerably to include more features, such as chronic pain, chronic fatigue, Dysautonomia, Mast Cell Activation Syndrome, Hypoglycemia, anxiety, and more.
ehlers-danlos.com
What are the Symptoms:
- constant subluxations and dislocations (joint laxity i.e. loose joints)
- Orthostatic intolerance (Dysautonomia)
- significant lightheadedness on standing, due to a slowed response by their circulatory system to compensation against blood pressure and flow changes with shifts in body position
- Bowel disorders especially functional dyspepsia (indigestion) and irritable bowel syndrome
- poor strength of collagen
- skin hyper-extensibility
- muscle pains and fatigue
- slow healing wounds
- fragile blood vessels
- TMJ- sore jaw/ pain with chewing
- serious nausea
- ganglion cysts i.e. rounded hard lumps in the wrists
- Gastroparesis
- Pelvis and Bladder Dysfunction
- Fatigue conditions
- Mast Cell Activation Syndrome
- Stiff joints and muscles/ joints that click
- Insomnia and unrefreshing sleep
- RSIs (Repetition Strain Injuries)
from rheumatologist Professor Rodney Grahame, healthline.com, ehlers-danlos.com, and rarediseases.org
Age and severity of onset vary quite widely from very mild (almost undetectable) to quite severe – from one individual to the next. Also, this genetic disorder can appear to skip a generation or a mutation can arise spontaneously, lending to the diagnostic confusion. Though some lucky people never have a major problem, all super-flexible people should take care and try to check so as to avoid any permanent injuries or dislocations over time. You can be fine one day, and then suddenly start to sublux or dislocate one or more joints more permanently, as well as developing arthritis in such loose joints. This is part of the reason it takes more than 10 years, on average, to be diagnosed…. since patients present with such a widely varying constellation of seemingly unrelated symptoms – everything from myopia to IBS, TMJ, hernias, migraines, low blood pressure, hemorrhoids and flat feet – until a doctor realizes the common collagen “thread” running through them all. EDS is often accompanied by symptoms of dysautonomia (low BP, poor temp regulation, Raynaud’s) to some degree, and may include POTS (postural orthostatic tachycardia syndrome) or NMH (neurally mediated hypotension), poor temperature regulation or GI motility issues.
chronicpainpartners.com
Are there treatment options for EDS?
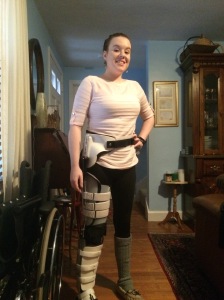
This is an HKAFO! I wear this whenever I am out and about to keep my hip from dislocating.
There is no specific cure for Ehlers-Danlos syndrome (EDS). The treatment and management is focused on preventing serious complications and relieving signs and symptoms. For example, physical therapy is often recommended to strengthen muscles and improve joint stability. Assistive devices such as braces, wheelchairs, or scooters may be necessary depending on the severity of joint instability. Pain medications may be prescribed to manage severe musculoskeletal (muscle and bone) pain. Affected people may be monitored for the development of osteopenia (low bone density) and aortic root dilatation (enlargement of the blood vessel that distributes blood from the heart to the rest of the body).
rarediseases.info.nih.gov
The Muldowney Clinic in RI has lots of information in their therapy program and if you can’t visit them everything is in their book “Living Life to the Fullest with Ehlers Danlos Syndrome.”
*none of this is medical advice and you should always talk to your doctor about any treatments you pursue.
